
GeneTree stable IDs in Compara
We provide stable IDs for some gene trees. These IDs are kept from one release to the other depending on the genes present in the Gene Tree. In particular, we do not rely on the underlying alignment, the tree structure or the resulting homologues to assign stable IDs to GeneTrees.
Format
The format of the stable IDs is ENSGTRRRRXXXXXXXXXX:
- Prefix ENSGT indicates that this is an Ensembl Vertebrates gene tree ID. Each row of the table below shows the Gene-Tree stable ID prefix associated with an Ensembl comparative genomics data resource.
- RRRR corresponds to the Ensembl release number when the stable ID was first assigned.
- XXXXXXXXXX is a unique number.
For instance, ENSGT00560000077204 is a gene tree first described in Ensembl 56.
Ensembl GeneTree stable ID prefixes
| Prefix | Comparative resource |
|---|---|
| EFGT0 | Fungi |
| EGGT0 | Pan-taxonomic compara |
| EMGT0 | Metazoa |
| ENSGT | Vertebrates |
| EPlGT | Plants |
| EPrGT | Protists |
Stable ID mapping
This is a generic description of the Stable ID mapping algorithm. We use it to track stable IDs from release to release, but we also use it to match Ensembl GeneTrees to TreeFam entries.
The algorithm takes two different classification schemes (i.e. Gene-Trees from the current release and Gene-Trees from the forthcoming one) over a given set of members. It is assumed that the classifications are somehow related, i.e. they tend to group members similarly. One is called "old" and the other "new", given an order of succession.
The comparison requires a common namespace for the members used in both classifications. The algorithm then infers how the names of the classes in two different classifications are related. To compare the Gene-Trees, we use the ENSEMBL translation stable_ids.
With respect to the two given classifications, we have three kinds of members:
- SHARED members (the ones present in both classifications)
- DISAPPEARING members (the ones present only in the "old" classification) - e.g. for the GeneTrees, gene predictions or complete genomes that disappear in the next release
- NEWBORN members (the ones present only in the "new" classification). - e.g. for the GeneTrees, new gene predictions or new complete genomes in the next release
The relationship between classes is inferred from the SHARED members, but the other two kinds are also counted by the algorithm.
The algorithm iterates through the "new" classes in the descending order of their sizes, trying to reuse a name of one of the "old" classes from where the SHARED members come to the "new" class, and make it the name of the "new" class, if it has not been taken yet.
If 100% SHARED members of the "old" class become 100% SHARED members of the new class, we call this case an EXACT reuse. If there was only one SHARED member, we call it an EXACT_o for "orphan".
Otherwise we have a split/join situation and iterate through the "contributors" (the "old" classes from which the SHARED members come from) in the decreasing order of the sizes of the shared parts. This ordering ensures that both in cases of joins and splits we are reusing the name of the biggest contributor.
- If the "old" class name of the first (the biggest) part has not yet been taken, we take it and call this a MAJORITY reuse. Due to the ordering (and looking at the statistics) this type of name reuse usually means that the SHARED majority of the biggest "old" contributor becomes the SHARED majority of the "new" class. If there was only one SHARED member, we call it a MAJORITY_o for "orphan", although this will rarely happen.
- If the "old" class name of the first part has been taken, but one of the other "old" class names is still available, we take it and call it a NEXTBEST name. (these classes are usually very small, because the majority of the SHARED members participate in EXACT/MAJORITY cases).
- If a name could not have been reused, because all the "old" contributor's names have been used, a completely new name is generated and it is a NEWNAME case. This usually means we are dealing with a split of a big class, where the majority has gone into a MAJORITY, and the rest of it needs a new name. Again, if there was only one SHARED member, we call it NEWNAME_o.
- Finally, if we have a "new" class that only contains NEWBORN members (meaning there were no "old" classes to reuse the names from), a new name is also generated, but this is a NEWFAM case. If the new class has only one (NEWBORN) member, it is a NEWFAM_o case.
In this example diagram:
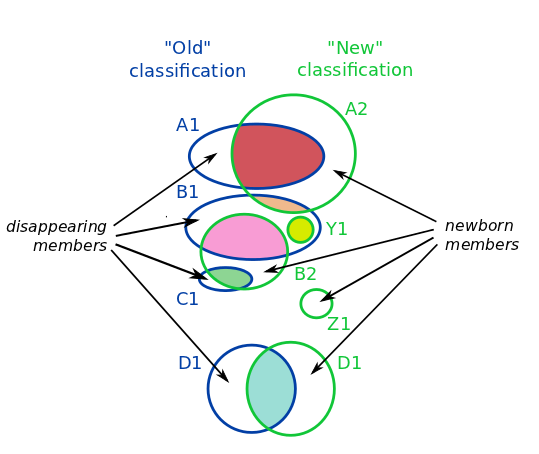
- D1 is an EXACT reuse (cyan contributor)
- A2 inherits the name from A1 according to the MAJORITY rule (light red contributor)
- B2 inherits the name from B1 (pink contributor), and C1 name disappears in the new classification and it is merged into B2 (light green).
- Z1 is created from NEWBORN members
- Y1 is an example of NEWNAME (yellow contributor)
Versioning
A version increase indicates that the SHARED members have changed for that class. The version is kept the same if it is an EXACT reuse case. For example, in the GeneTrees:
a) If a genetree with 50 members in release 56 turns into a genetree with 49+2 members in release 57, 49 being SHARED and 2 being NEWBORN, the version will change.
b) If a genetree with 50 members in release 56 turns into a genetree with 48+2 members in release 57, 48 being SHARED and 2 being NEWBORN, the version will change, even though the total number of members is the same.
c) If a genetree with 48+2 members in release 56 turns into a genetree with 48 members in release 57, 48 being SHARED and 2 being DISAPPEARING (e.g. updated genebuild that deletes some members), the version will be the same.
d) If a genetree with 48+2 members in release 56 turns into a genetree with 48+3 members in release 57, 48 being SHARED, 2 being DISAPPEARING and 3 being NEWBORN, the version will be the same.