
Orthology quality-controls
We have two methods to provide quality scores for orthologue pairs:
These methods are completely indepenent of each other and of the orthology inference itself. The scores can be used to determine how likely it is that the orthologue pairs are real.
Gene Order Conservation score
Genes that are descended from the same gene are likely to be part of a block of genes, all in the same order, in both species. Some rearrangements between genes may occur over time, particularly in distantly related species, but it is less likely that an isolated gene which does not share gene neighbours with its inferred orthologue is a real orthologue.
The gene order conservation (GOC) score indicates how many of the four closest neighbours of a gene match between orthologous pairs. It is calculated by following these steps:
- Load all the predicted orthologues for a pair of species
- Separate the orthologues into their respective chromosomes
- Discard any orthologue that is by itself (usually in a scaffold). As these orthologues automatically get a NULL score for having no neighbours
- Order the set of orthologues in each chromosome by their start positions on the chosen reference genome
- For each orthologous pair, fetch the two genes upstream and downstream
- Check whether they are also identified as orthologues and in the same orientation
- Each match is scored as 25% meaning if all four neighbouring genes match that orthologue gets a GOC score of 100% for this reference genome
- Go back to step 4 and repeat using the alternative species as the reference genome
- Now we have two GOC scores for each other. We currently report the max of these scores
Example comparison
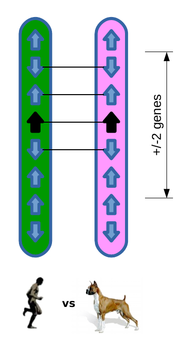
Of the four neighbouring genes, three are orthologues and in conserved order and position, resulting in a GOC score of 75.
Availability
Whole Genome Alignment score
We assume that genes which are orthologous to each other will fall within genomic regions that can be aligned to one another. Since we calculate pairwise whole genome alignments (WGA), we can use these to check the regions surrounding orthologues.
The whole genome alignment score calculates the coverage of the alignment over the orthologue pair, as follows:
- Exon boundaries are fetched for all genes in all species of interest
- The species are paired off and all alignments between each pair are detected. All predicted orthologues between the pair are fetched and batched (default = 10)
- The coverage over each member of the orthology is calculated using every available alignment. Coverage over exons is regarded as a higher importance than intronic regions, so a weighted score is generated. The score takes the coverage over exons as a base, with bonus points given for coverage over the introns (normalised by the proportion of intronic sequence in the gene).
- An overall score for the homology prediction, as a whole is computed. This can be defined as the maximum score, after the score for the pair of genes has been averaged for each alignment i.e. we report the average score for the greatest-coverage alignment
Example comparison

Availability
High-confidence orthologies
In every orthology inference between two species, the set of orthologue pairs between the species is considered for high-confidence annotation. The GOC and WGA scores among others are used to determine which orthologies to tag as being high-confidence.
The primary filter used to select the high-confidence set of orthologue pairs, consists of three thresholds for respectively the GOC score, the WGA coverage and the percentage identity of the orthologue pairs. The thresholds we use depend on the most recent common ancestor of the species pair, according to the table below. The primary filter is used if there are scores of either type (GOC or WGA) in the set of orthologues between the two species. The orthology between two genes will be classified as high confidence if the alignment between the two genes satisfies the percentage identity requirement and either GOC or WGA scores meet their thresholds.
If GOC or WGA scores are not available or if no threshold is used for a set of orthologues, the pipeline will use fallback criteria, that is the prediction will be classified as high confidence if the percentage identity meets the threshold and the prediction is tree compliant.
| Clades | Min. GOC score | Min. WGA score | Min. %identity |
|---|---|---|---|
| Apes, Murinae | 75 | 75 | 80 |
| Mammalia, Aves, Percomorpha | 75 | 75 | 50 |
| Others | 50 | 50 | 25 |