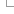

Ensembl Variant Effect Predictor Input form
When you reach the Ensembl VEP web interface, you will be presented with a form to enter your data and alter various options.
Note that the listed options change depending on the selected species.
Data input
- First select the correct species for your data. Ensembl hosts many vertebrate genomes; genomes for plants, protists and fungi can be found at Ensembl Genomes.
- You can optionally choose a name for the data you upload - this can make it easier for you to identify jobs and files that you have uploaded to the Ensembl VEP at a later point.
-
You have three options for uploading your data:
- File upload - click the "Choose file" button and locate the file on your system. Please ensure your files are sorted by location if you are using VCF or other input formats using location. This greatly improves speed of analysis
- Paste file - simply copy and paste the contents of your file into the large text box
- File URL - point the Ensembl VEP to a file hosted on a publically accessible address. This can be either a http:// or ftp:// address.
The format of your data is automatically detected; see the examples or the input format documentation.
- For pasted data you can get an instant preview of the results of your first variant by clicking the button that appears when you paste your data. This quickly shows you the consequence type, the IDs of any overlapping variants, genes, transcripts and regulatory features, as well as SIFT and PolyPhen predictions. To see the full results set submit your job as normal.
-
For some species you can select which transcript database to use. The default is to use Ensembl transcripts, which offer the most rich annotation through Ensembl VEP.
GENCODE Basic is a subset of the GENCODE gene set, and is intended to provide a simplified, high-quality subset of the GENCODE transcript annotations that will be useful to the majority of users. GENCODE Basic includes all genes in the GENCODE gene set, with a representative subset of the transcripts (splice variants).
GENCODE Primary is a new transcript subset which covers all human exons in a minimal set of transcripts. This aims to enable annotation of all potential variant consequences without duplication across multiple transcripts.
You can also select to use RefSeq transcripts from the otherfeatures database; note though that these transcripts are simply aligned to the reference genome and the database is missing much of the annotation found when using the main Ensembl database (e.g. protein domains, CCDS identifiers).
Identifiers
- Gene symbol
Add the gene symbol for the gene to the output. This will typically be, for example, the HGNC identifier for genes in human. Equivalent to --symbol in the Ensembl VEP script.
- Transcript version
Add the transcript version to the transcript identifier. Equivalent to --transcript_version.
- CCDS
Add the Consensus CDS transcript identifier where available. Equivalent to --ccds.
- Protein
Add the Ensembl protein identifer (ENSP). Equivalent to --protein.
- UniProt
Add identifiers for translated protein products from three UniProt-related databases (SWISSPROT, TREMBL and UniParc). Equivalent to --uniprot.
- HGVS
Generate HGVS identifiers for your input variants relative to the transcript coding sequence (HGVSc) and the protein sequence (HGVSp). Equivalent to --hgvs.
Variants and frequency data
-
Find co-located known variants - report known variants from the Ensembl Variation database that overlap with your input. A list of variant sources imported can be viewed here. Note that this feature is only available for species with an Ensembl Variation database. Equivalent to --check_existing.
Ensembl VEP will by default compares the alleles of your input variant to that of the existing variant; Ensembl VEP will only report the existing variant ID if none of the alleles in your input variant are novel.
For example, if your input variant has alleles A/G, and the existing variant has alleles A/T, then the existing variant will not be reported. If instead your input variant has alleles A/T, then the existing variant will be reported.
To disable this allele matching, select the option "Yes but don't compare alleles" for the option "Find co-located known variants".
For known variants Ensembl VEP can also provide PubMed IDs of publications citing the variant (equivalent to --pubmed).
- Variant synonyms
Report known synonyms for co-located variants.
- Frequency data for co-located variants
Ensembl VEP can also report allele frequency (AF) data for existing variants from several major genotyping projects, the 1000 Genomes Project, and gnomAD; this only applies when you have selected human as your species.
- 1000 Genomes global - the combined phase 3 population (i.e. all individuals from all populations). Equivalent to --af
- 1000 Genomes continental - the four continent-level populations - AFR (African), AMR (American), ASN (Asian) and EUR (European). Equivalent to --af_1kg
- gnomAD exomes - combined, AFR, AMR, ASJ, EAS, FIN, NFE, OTH, SAS populations. Equivalent to --af_gnomade
- gnomAD genomes - combined, AFR, AMR, AMI, ASJ, EAS, FIN, MID, NFE, OTH, SAS populations. Equivalent to --af_gnomadg
- PubMed IDs for citations of co-located variants
Report the PubMed IDs of any publications that cite the co-located variant(s).
- Include flagged variants
Variants flagged as failed by the Ensembl Variation quality control.
- Paralogue variants
Retrieves variants that overlap genomic coordinates corresponding to aligned amino acid positions in paralogous proteins. This functionality is provided by the Paralogues plugin.
- Open Targets Platform
Report trait associations from Open Targets Platform through GWAS- or QTL-gene associations, including locus-to-gene (L2G) scores for GWAS loci Open Targets Platform. This functionality is provided by the OpenTargets plugin.
Additional annotations
- DosageSensitivity
Retrieves haploinsufficiency and triplosensitivity probability scores for affected genes from a dosage sensitivity catalogue published in paper - https://www.sciencedirect.com/science/article/pii/S0092867422007887.
- LOEUF
LOEUF stands for the 'loss-of-function observed/expected upper bound fraction'. This plugin adds constraint scores derived from gnomAD to Ensembl VEP. Equivalent to the Ensembl VEP plugin LOEUF.
- Transcript biotype
Add the transcript biotype to the output. Equivalent to --biotype in the Ensembl VEP script.
- Exon and intron numbers
Report the exon or intron number that a variant falls in as NUMBER / TOTAL, i.e. exon 2/5 means the variant falls in the 2nd of 5 exons in the transcript. Equivalent to --numbers.
- Transcript support level
Report the transcript support level of the overlapped transcript. Equivalent to --tsl.
- APPRIS
Report the APPRIS score of the overlapped transcript. Equivalent to --appris.
- Identify canonical transcripts
Add a flag to the output indicating if the reported transcript is the canonical transcript for the gene. Equivalent to --canonical.
- Upstream/Downstream distance (bp)
Change the distance to assign the upstream and downstream consequences. Equivalent to --distance.
- miRNA structure
Determines where in the secondary structure of a miRNA a variant falls (only for Ensembl/GENCODE transcripts). Equivalent to --mirna.
- NMD
Predicts if a stop_gained variant allows the transcript to escape nonsense-mediated mRNA decay based on certain rules. This functionality is provided by the NMD plugin.
- UTRAnnotator
Annotates the effect of 5' UTR variant especially for variant creating/disrupting upstream ORFs. This functionality is provided by the UTRAnnotator plugin.
- RiboseqORFs
Annotates the consequences of variants overlapping Ribo-seq ORFs. This functionality is provided by the RiboseqORFs plugin.
- Protein matches
Shows the variant location on PDBe and AlphaFold protein structures in interactive 3d displays, where available. Report protein domains from PDBe, Pfam, Prosite and InterPro that overlap input variants. Equivalent to --domains.
- mutfunc
mutfunc predicts destabilization effect of protein structure, interaction, regulatory region, etc. caused by a variant. This functionality is provided by the mutfunc plugin.
- ProtVar
ProtVar ProtVar provides contextualised information for missense variation, including destabilisation of protein structures, overlapping protein pockets, and protein-protein interaction interfaces. This functionality is provided by the ProtVar plugin.
- IntAct
Reports relevant data for variants that falls within molecular interaction site as reprted by IntAct database. This functionality is provided by the IntAct plugin.
- MaveDB
MaveDB holds experimentally determined measures of variant effect. This functionality is provided by the MaveDB plugin.
- Get regulatory region consequences
In addition to predicting consequences with overlapping transcripts, Ensembl VEP can find overlaps with known regulatory regions as determined in the Ensembl Regulatory build.
Using this option, Ensembl VEP will also report if a variant falls in a transcription factor binding motif, and give a score that reflects whether the altered motif sequence is more or less similar to the consensus.
Get regulatory consequences is equivalent to --regulatory. - Enformer
Predictions of variant impact on gene expression. This functionality is provided by the Enformer plugin.
- Phenotypes
Report the phenotypic data overlapping the genomic features. This functionality is provided by the Phenotypes plugin.
For more information on the imported phenotypic data for genes, variation and QTLs see our phenotype documentation.
Note: This web functionality is not reporting cancer phenotypic data this release. However the cancer phenotypic data is available in the command line version. - Gene Ontology
Add terms to describe any overlapping gene's function, the cellular component in which the function is performed and the biological processes to which this contributes.
- Geno2MP
Geno2MP is a web-accessible database of rare variant genotypes linked to individual-level phenotypic profiles defined by human phenotype ontology (HPO) terms. This functionality is provided by the Geno2MP plugin.
- Mastermind
Uses the Mastermind Genomic Search Engine to report variants that have clinical evidence cited in the medical literature. This functionality is provided by the Mastermind plugin.
Note: This web functionality is only reporting the URL to the Mastermind Genomic Search Engine webpage.
Predictions
- SIFT predictions
SIFT predicts whether an amino acid substitution affects protein function based on sequence homology and the physical properties of amino acids. Only available in popular species. For both SIFT and PolyPhen Ensembl VEP can report either a score between 0 and 1, a prediction in words, or both. Equivalent to --sift.
- PolyPhen predictions
PolyPhen is a tool which predicts possible impact of an amino acid substitution on the structure and function of a human protein using straightforward physical and comparative considerations. Equivalent to --polyphen.
- dbNSFP
Retrieves data for missense variants from dbNSFP. Equivalent to the Ensembl VEP plugin dbNSFP.
- AlphaMissense
AlphaMissense is a deep learning model developed by Google DeepMind that predicts the pathogenicity of single nucleotide missense variants. Equivalent to the Ensembl VEP plugin AlphaMissense.
- CADD
Combined Annotation Dependent Depletion (CADD) is a tool for scoring the deleteriousness of single nucleotide variants and insertion/deletion variants in the human genome. CADD integrates multiple annotations into one metric by contrasting variants that survived natural selection with simulated mutations. CADD is only available here for non-commercial use. See CADD website for more information. Equivalent to the Ensembl VEP plugin CADD.
- REVEL
Rare Exome Variant Ensemble Learner (REVEL) is an ensemble method for predicting the pathogenicity of missense variants based on a combination of scores from multiple individual tools. REVEL is only available here for non-commercial use. Equivalent to the Ensembl VEP plugin REVEL.
- ClinPred
ClinPred is a prediction tool to identify disease-relevant nonsynonymous single nucleotide variants. The predictor incorporates existing pathogenicity scores and benefits from normal population allele frequencies. ClinPred is only available here for non-commercial use. Equivalent to the Ensembl VEP plugin ClinPred.
- EVE
Adds information from EVE (evolutionary model of variant effect). Equivalent to the Ensembl VEP plugin EVE.
- dbscSNV
Retrieves data for splicing variants from dbscSNV. Equivalent to the Ensembl VEP plugin dbscSNV.
- MaxEntScan
Get splice site predictions from MaxEntScan. Equivalent to the Ensembl VEP plugin MaxEntScan.
- SpliceAI
Pre-calculated annotations from SpliceAI a deep neural network, developed by Illumina, Inc that predicts splice junctions from an arbitrary pre-mRNA transcript sequence. Used for non-commercial purposes. This functionality is provided by the SpliceAI plugin.
The pre-calculated annotations for all possible single nucleotide substitutions can be retrieved from:- Ensembl/GENCODE v24 canonical transcripts
Masked scores
- Ensembl/GENCODE v37 MANE transcripts
Raw scores
- Ensembl/GENCODE v24 canonical transcripts
- BLOSUM62
Looks up the BLOSUM 62 substitution matrix score for the reference and alternative amino acids predicted for a missense mutation. Equivalent to the Ensembl VEP plugin Blosum62.
- Ancestral allele
Retrieves ancestral allele sequences from a FASTA file. Ensembl produces FASTA file dumps of the ancestral sequences of key species. Equivalent to the Ensembl VEP plugin AncestralAllele.
Filtering options
- By frequency
Filter variants by minor allele frequency (MAF). Two options are provided:
- Exclude common variants
Filter out variants that are co-located with an existing variant that has a frequency greater than 0.01 (1%) in the 1000 Genomes global population. Equivalent to --filter_common in the Ensembl VEP script.
- Advanced filtering
Enabling this option allows you to specify a population and frequency to compare to, as well whether matching variants should be included or excluded from the results.
- Exclude common variants
- Return results for variants in coding regions only
Exclude variants that don't fall in a coding region of a transcript. Equivalent to --coding_only.
- Restrict results
For many variants Ensembl VEP will report multiple consequence types - typically this is because the variant overlaps more than one transcript. For each of these options Ensembl VEP uses consequence ranks that are subjectively determined by Ensembl. This table gives all of the consquence types predicted by Ensembl, ordered by rank. Note that enabling one of these options not only loses potentially relevant data, but in some cases may be scientifically misleading. Options:
- Show one selected consequence
Pick one consequence type across all those predicted for the variant; the output will include transcript- or feature-specific information. Consequences are chosen by the canonical, biotype status and length of the transcript, along with the ranking of the consequence type according to this table. This is the best method to use if you are interested only in one consequence per variant. Equivalent to --pick.
- Show one selected consequence per gene
Pick one consequence type for each gene using the same criteria as above. Note that if a variant overlaps more than one gene, output for each gene will be reported. Equivalent to --per_gene.
- Show only list of consequences per variant
Give a comma-separated list of all observed consequence types for each variant. No transcript-specific or gene-specific output will be given. Equivalent to --summary.
- Show most severe per variant
Only the most severe of all observed consequence types is reported for each variant. No transcript-specific or gene-specific output will be given. Equivalent to --most_severe.
- Show one selected consequence
Advanced options
The Ensembl VEP web interface allows you to use/setup advanced options:
- Buffer size
By default Ensembl VEP process the variants by blocks of 5000 (i.e. what we call "buffer size").
In some cases, reducing the size of the blocks (buffer size) could prevent memory issues for large Ensembl VEP queries (e.g. use of regulatory data, many plugins or custom annotations).
This is why the maximum buffer size is automatically set to 500 on the Ensembl VEP Web interface when the "Regulatory data" option is selected. - Right align variants prior to consequence calculation
By default Ensembl VEP performs consequence calculation at the given input coordinates.
Optionally, Ensembl VEP can shift insertions and deletions found within repeated regions as far as possible in the 3' direction, normalising output.
Jobs
Once you have clicked Run, your input will be checked and submitted to the Ensembl VEP as a job. All jobs associated with your session or account are shown in the Recent Tickets table. You may submit multiple jobs simultaneously.
The Jobs column of the table shows the current status of the job.
- Queued - your job is waiting to be submitted to the system
- Running - your job is currently running
- Done - your job is finished - click the [View results] link to be taken to the results page
- Failed
- there is a problem with your job - click the magnifying glass icon
to see more details
- Save icon: save the job (you
need to login with an Ensembl account).
- Edit icon: resubmit a job
(for example, to slightly tweak the data or parameters before
re-running).
- Magnifying glass icon: see
summary of the options that you selected for your Ensembl VEP job, as well
as data versions associated with this run.
- Share icon: display URL to
share with other users. You can also disable URL sharing here.
- Trash can icon: delete a
job.
| Analysis | Jobs | Submitted at (GMT) | |
|---|---|---|---|
| Ensembl Variant Effect Predictor |  Ensembl VEP analysis of pasted data in Bos_taurusDone[View results] Ensembl VEP analysis of pasted data in Bos_taurusDone[View results] | 2023051117004011/05/2023, 17:00 | |
| Ensembl Variant Effect Predictor |  Ensembl VEP analysis of pasted data in Ovis_ariesDone[View results] Ensembl VEP analysis of pasted data in Ovis_ariesDone[View results] | 2023051116553211/05/2023, 16:55 | |
| Ensembl Variant Effect Predictor |  Ensembl VEP analysis of pasted data in Homo_sapiensFailed Ensembl VEP analysis of pasted data in Homo_sapiensFailed | 2023042009570511/05/2023, 16:54 | |
| Ensembl Variant Effect Predictor | Ensembl VEP analysis of pasted data in Homo_sapiensRunning | 2023042009570511/05/2023, 16:51 | |
| Ensembl Variant Effect Predictor | Ensembl VEP analysis of pasted data in Homo_sapiensQueued | 2023042009570511/05/2023, 16:49 |